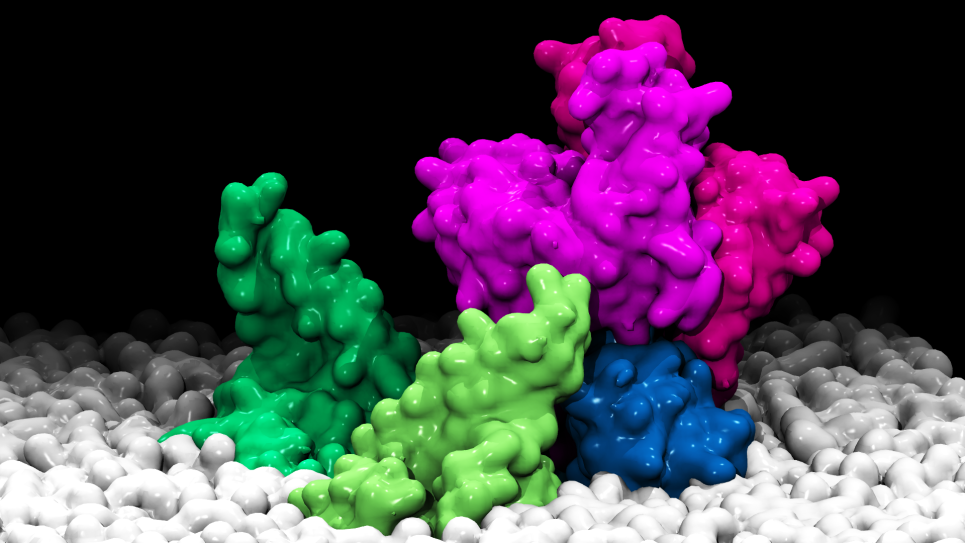
Improvement in predicting the ability of small molecules to bind to proteins can further basic knowledge in human health and holds the promise for improved processes in drug discovery. However, the success rate for docking has been mixed historically. Many of the shortcomings point to the scoring functions and the approximations and heuristics necessary to make the run-time feasible. The vast computing resources available now remove some of these constraints, allowing more advanced physics-based methods to be studied. Implementing these methods and making them more accessible to researchers helps to realize more of the promise that molecular simulation holds.
Using the advanced Blue Gene/P system, researchers are conducting a comprehensive analysis of protein binding domain and small molecule interactions through an automated system, including receptor analysis, protein-ligand docking, and binding free energy calculations. In addition, the team is performing the first, large-scale study of the computationally intensive FEP/MD-GCMC methodology for estimating free binding energy. Finally, in collaboration with the Center for Structural Genomics of Infectious Diseases, the team is conducting computer-aided drug discovery on human pathogens. The predicted computational results will be experimentally tested in binding assays and X-ray crystallography experiments, allowing for the important validation step necessary to evaluate the predictive power of biomolecular simulations.
In the next phase of the project, the team will use a production code to run protein-ligand interaction simulations on human pathogen targets of high biomedical value, such as S. aureus and B. anthracis. During 2012, the team will continue to operate the pipeline as originally designed regarding protein targets being studied. However, the team will begin to implement FEP calculations using NAMD, replacing the CHARMM-based calculations previously employed. They will also continue to work with biochemists and protein crystallographers to validate the computational predictions from their pipeline.